Plots the structure of specified path found by pathClassifier.
Source:R/pathClassifier.R
plotPathClassifier.RdPlots the structure of specified path found by pathClassifier.
plotPathClassifier(ybinpaths, obj, m, tol = NULL)Arguments
- ybinpaths
The training paths computed by
pathsToBinary- obj
The pathClassifier
pathClassifier.- m
The path component to view.
- tol
A tolerance for 3M parameter
thetawhich is the probability for each edge within each cluster. If the tolerance is set all edges with athetabelow that tolerance will be removed from the plot.
Value
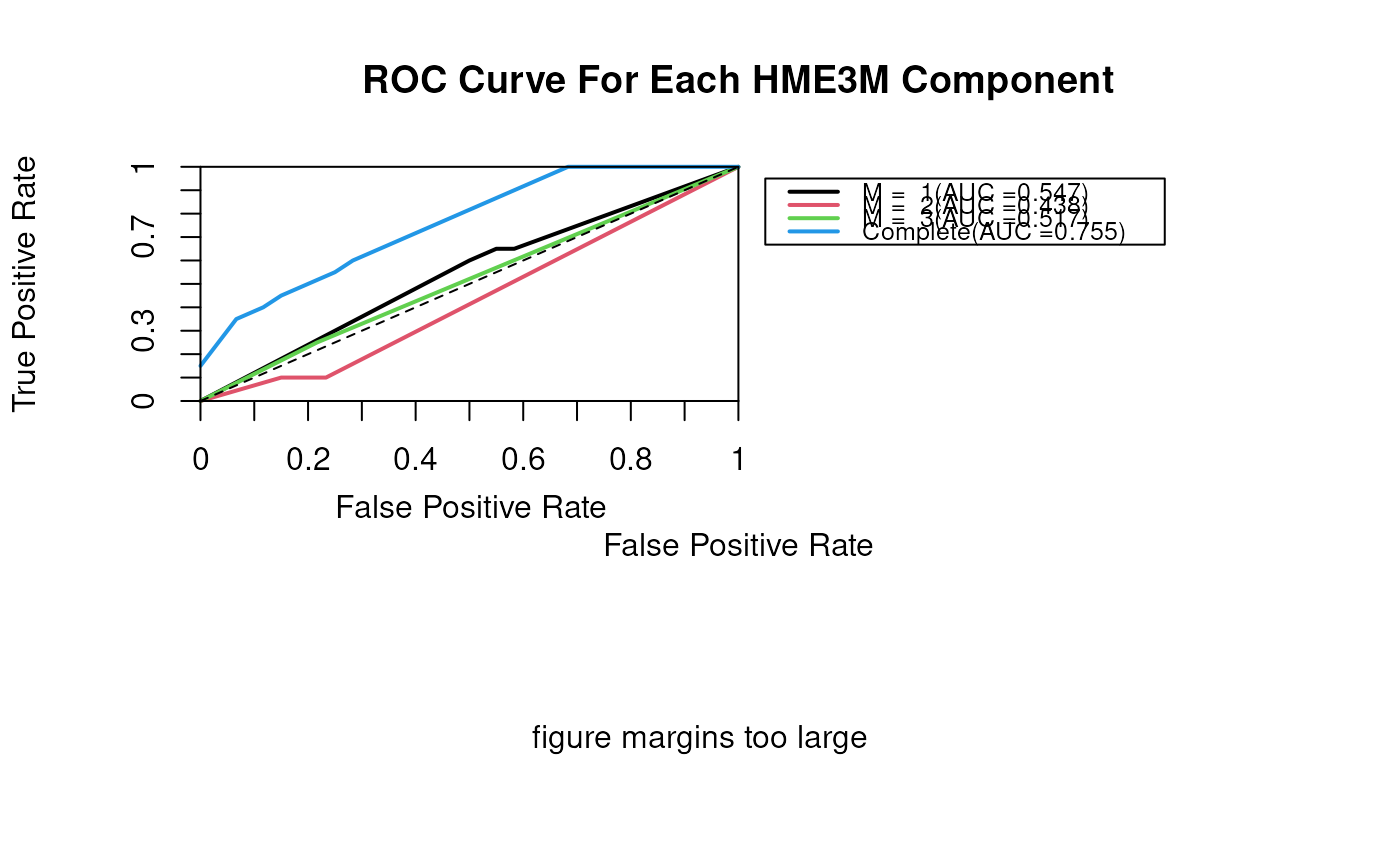
Produces a plot of the paths with the path probabilities and prediction probabilities and ROC curve overlayed.
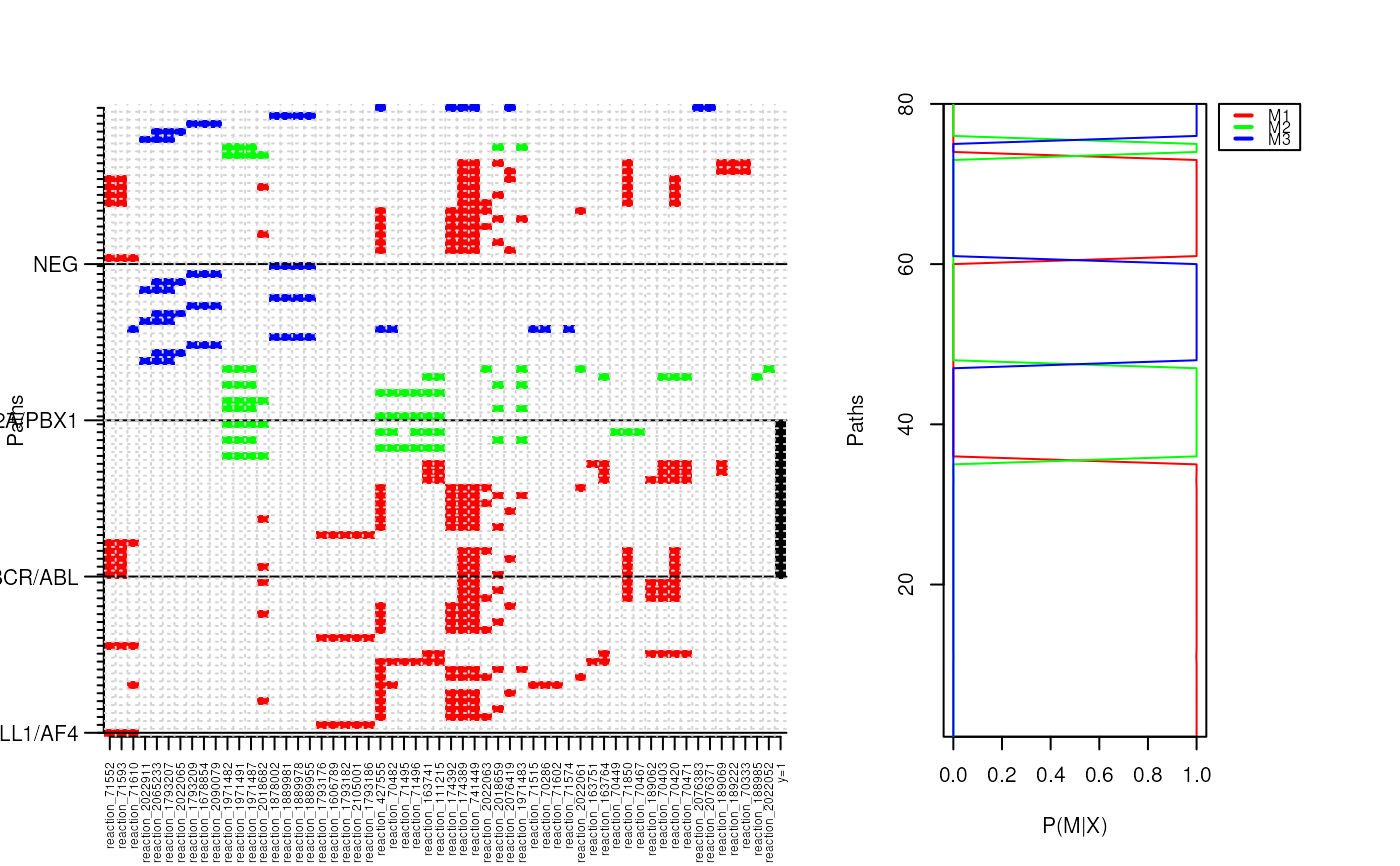
- Center Plot
An image of all paths the training dataset. Rows are the paths and columns are the genes (vertices) included within each pathway. A colour within image indicates if a particular gene (vertex) is included within a specific path. Colours flag whether a path belongs to the current HME3M component (P > 0.5).
- Center Right
The training set posterior probabilities for each path belonging to the current 3M component.
- Center Top
The ROC curve for this HME3M component.
- Top Bar Plots
Theta: The 3M component probabilities - indicates the importance of each edge is to a path.Beta: The PLR coefficient - the magnitude indicates the importance of the edge to the classify the response.
See also
Other Path clustering & classification methods:
pathClassifier(),
pathCluster(),
pathsToBinary(),
plotClassifierROC(),
plotClusterMatrix(),
plotPathCluster(),
predictPathClassifier(),
predictPathCluster()
Other Plotting methods:
colorVertexByAttr(),
layoutVertexByAttr(),
plotAllNetworks(),
plotClassifierROC(),
plotClusterMatrix(),
plotCytoscapeGML(),
plotNetwork(),
plotPaths()
Examples
## Prepare a weighted reaction network.
## Conver a metabolic network to a reaction network.
data(ex_sbml) # bipartite metabolic network of Carbohydrate metabolism.
rgraph <- makeReactionNetwork(ex_sbml, simplify=TRUE)
#> This graph was created by an old(er) igraph version.
#> ℹ Call `igraph::upgrade_graph()` on it to use with the current igraph version.
#> For now we convert it on the fly...
## Assign edge weights based on Affymetrix attributes and microarray dataset.
# Calculate Pearson's correlation.
data(ex_microarray) # Part of ALL dataset.
rgraph <- assignEdgeWeights(microarray = ex_microarray, graph = rgraph,
weight.method = "cor", use.attr="miriam.uniprot",
y=factor(colnames(ex_microarray)), bootstrap = FALSE)
#> 100 genes were present in the microarray, but not represented in the network.
#> 55 genes were couldn't be found in microarray.
#> Assigning edge weights for label ALL1/AF4
#> Assigning edge weights for label BCR/ABL
#> Assigning edge weights for label E2A/PBX1
#> Assigning edge weights for label NEG
## Get ranked paths using probabilistic shortest paths.
ranked.p <- pathRanker(rgraph, method="prob.shortest.path",
K=20, minPathSize=6)
#> Extracting the 20 most probable paths for ALL1/AF4
#> Extracting the 20 most probable paths for BCR/ABL
#> Extracting the 20 most probable paths for E2A/PBX1
#> Extracting the 20 most probable paths for NEG
## Convert paths to binary matrix.
ybinpaths <- pathsToBinary(ranked.p)
p.class <- pathClassifier(ybinpaths, target.class = "BCR/ABL", M = 3)
## Plotting the classifier results.
plotClassifierROC(p.class)
 plotClusters(ybinpaths, p.class)
plotClusters(ybinpaths, p.class)