HME3M Markov pathway classifier.
pathClassifier(
paths,
target.class,
M,
alpha = 1,
lambda = 2,
hme3miter = 100,
plriter = 1,
init = "random"
)Arguments
- paths
The training paths computed by
pathsToBinary- target.class
he label of the targe class to be classified. This label must be present as a label within the
paths\$yobject- M
Number of components within the paths to be extracted.
- alpha
The PLR learning rate. (between 0 and 1).
- lambda
The PLR regularization parameter. (between 0 and 2)
- hme3miter
Maximum number of HME3M iterations. It will stop when likelihood change is < 0.001.
- plriter
Maximum number of PLR iteractions. It will stop when likelihood change is < 0.001.
- init
Specify whether to initialize the HME3M responsibilities with the 3M model - random is recommended.
Value
A list with the following elements. A list with the following values
- h
A dataframe with the EM responsibilities.
- theta
A dataframe with the Markov parameters for each component.
- beta
A dataframe with the PLR coefficients for each component.
- proportions
The probability of each HME3M component.
- posterior.probs
The HME3M posterior probability.
- likelihood
The likelihood convergence history.
- plrplr
The posterior predictions from each components PLR model.
- path.probabilities
The 3M probabilities for each path belonging to each component.
- params
The parameters used to build the model.
- y
The binary response variable used by HME3M. A 1 indicates the location of the target.class labels in
paths\$y- perf
The training set ROC curve AUC.
- label
The HME3M predicted label for each path.
- component
The HME3M component assignment for each path.
Details
Take care with selection of lambda and alpha - make sure you check that the likelihood is always increasing.
References
Hancock, Timothy, and Mamitsuka, Hiroshi: A Markov Classification Model for Metabolic Pathways, Workshop on Algorithms in Bioinformatics (WABI) , 2009
Hancock, Timothy, and Mamitsuka, Hiroshi: A Markov Classification Model for Metabolic Pathways, Algorithms for Molecular Biology 2010
See also
Other Path clustering & classification methods:
pathCluster(),
pathsToBinary(),
plotClassifierROC(),
plotClusterMatrix(),
plotPathClassifier(),
plotPathCluster(),
predictPathClassifier(),
predictPathCluster()
Examples
## Prepare a weighted reaction network.
## Conver a metabolic network to a reaction network.
data(ex_sbml) # bipartite metabolic network of Carbohydrate metabolism.
rgraph <- makeReactionNetwork(ex_sbml, simplify=TRUE)
#> This graph was created by an old(er) igraph version.
#> ℹ Call `igraph::upgrade_graph()` on it to use with the current igraph version.
#> For now we convert it on the fly...
## Assign edge weights based on Affymetrix attributes and microarray dataset.
# Calculate Pearson's correlation.
data(ex_microarray) # Part of ALL dataset.
rgraph <- assignEdgeWeights(microarray = ex_microarray, graph = rgraph,
weight.method = "cor", use.attr="miriam.uniprot",
y=factor(colnames(ex_microarray)), bootstrap = FALSE)
#> 100 genes were present in the microarray, but not represented in the network.
#> 55 genes were couldn't be found in microarray.
#> Assigning edge weights for label ALL1/AF4
#> Assigning edge weights for label BCR/ABL
#> Assigning edge weights for label E2A/PBX1
#> Assigning edge weights for label NEG
## Get ranked paths using probabilistic shortest paths.
ranked.p <- pathRanker(rgraph, method="prob.shortest.path",
K=20, minPathSize=6)
#> Extracting the 20 most probable paths for ALL1/AF4
#> Extracting the 20 most probable paths for BCR/ABL
#> Extracting the 20 most probable paths for E2A/PBX1
#> Extracting the 20 most probable paths for NEG
## Convert paths to binary matrix.
ybinpaths <- pathsToBinary(ranked.p)
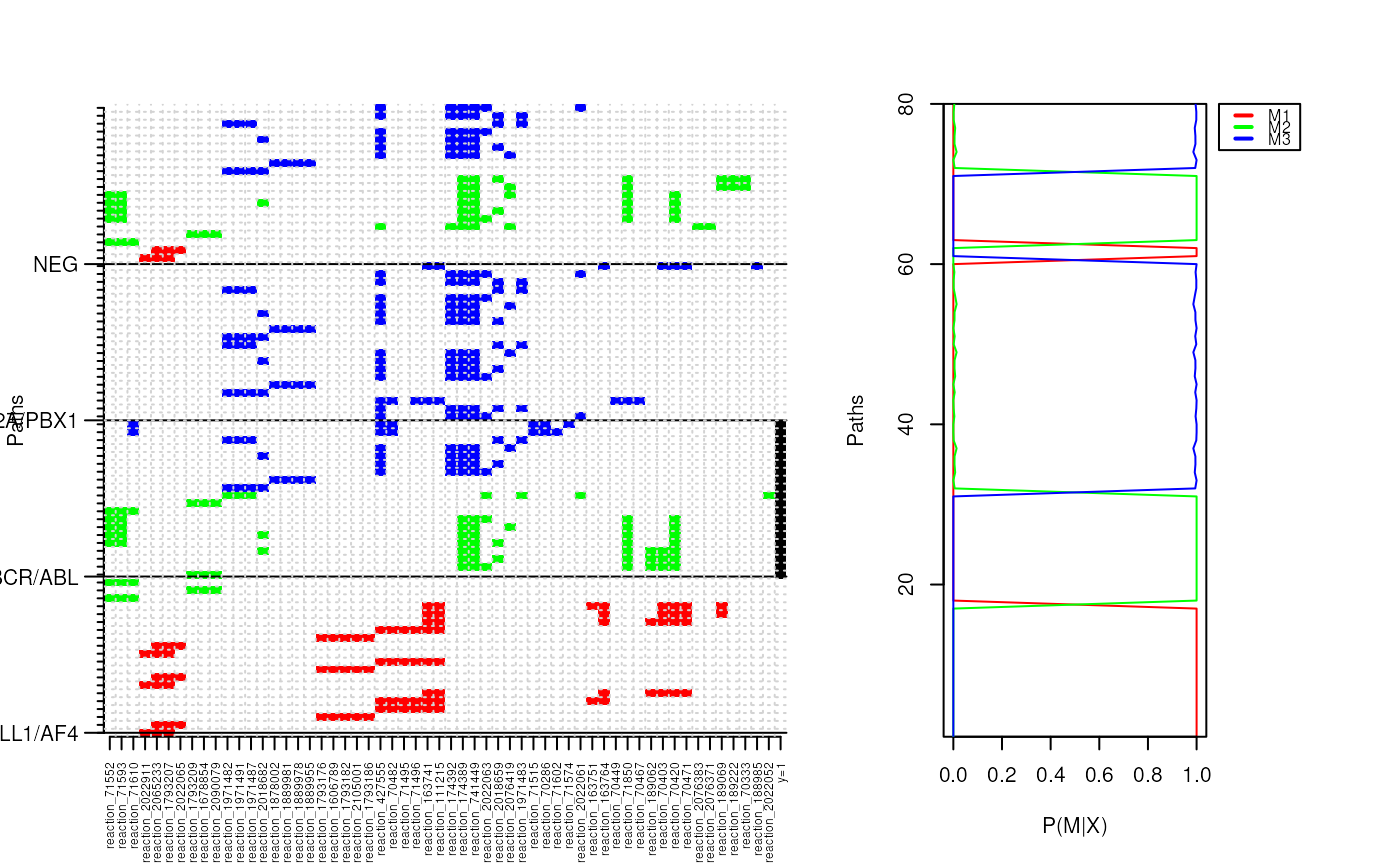
p.class <- pathClassifier(ybinpaths, target.class = "BCR/ABL", M = 3)
## Contingency table of classification performance
table(ybinpaths$y,p.class$label)
#>
#> 0 1
#> ALL1/AF4 20 0
#> BCR/ABL 16 4
#> E2A/PBX1 19 1
#> NEG 20 0
## Plotting the classifier results.
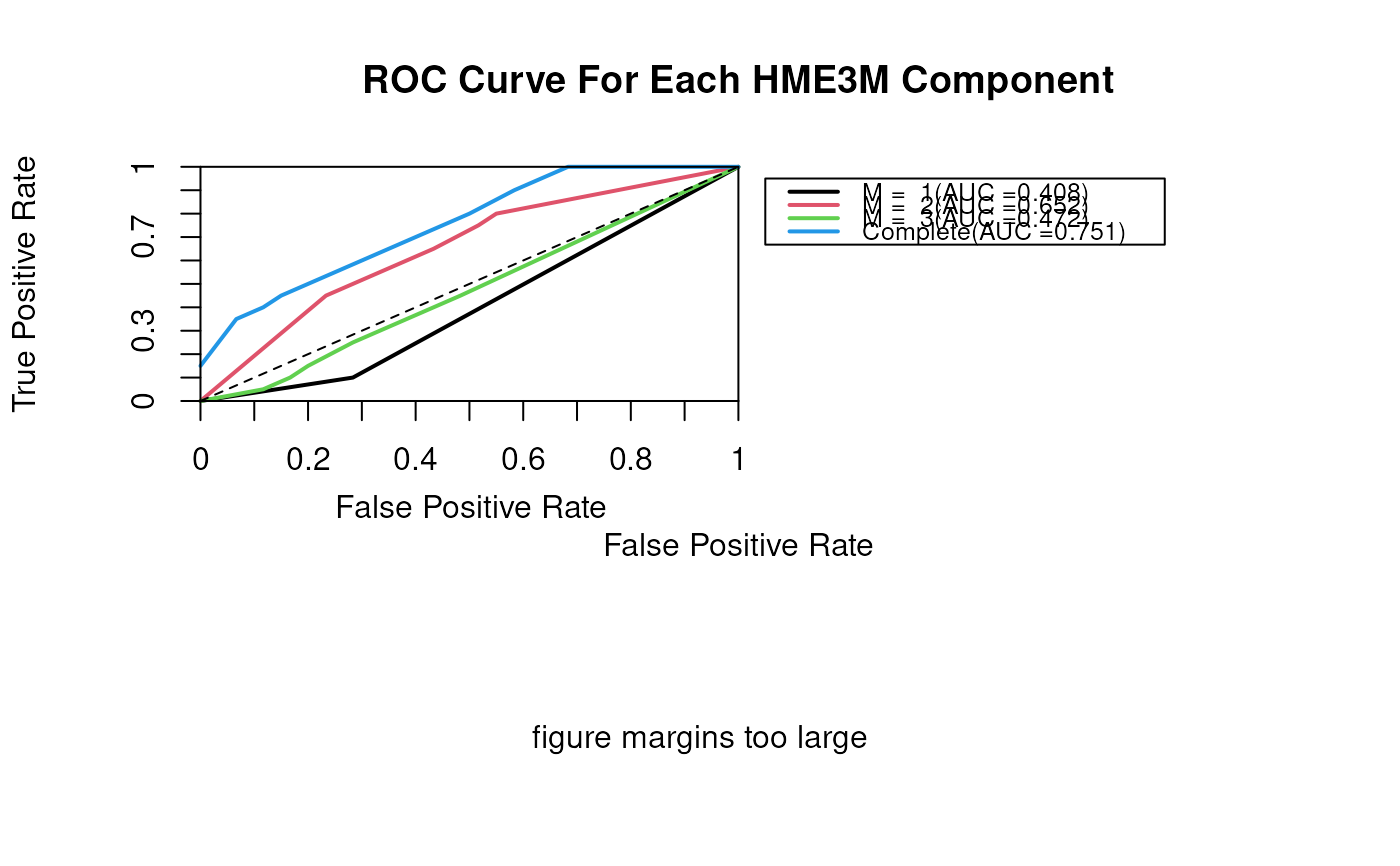
plotClassifierROC(p.class)
 plotClusters(ybinpaths, p.class)
plotClusters(ybinpaths, p.class)